
Resources For Researchers and Clinicians
Osteogenesis Imperfecta (OI) is a heterogeneous group of disorders that results in brittle bone due to a variety of problems with type I collagen including haploinsufficient type I collagen OI, dominant negative type I collagen mutation OI, disorders of type I collagen post-translational modification, disorders of type I collagen transport, and disorders of matrix cell signaling. These different mechanisms are anticipated to result in differences in phenotype, long-term outcome and responses to current and emerging therapies employed in the treatment of OI. Brittle bone disease or OI affects an estimated six to seven per 100,000 people worldwide. The number of Americans with OI is thought to be 25,000-50,000. Patients present with recurrent bone fractures due to bone fragility. Other variable features of OI include hearing loss, scoliosis, craniofacial abnormalities, short stature, dentinogenesis imperfecta, ligamentous laxity and blue-gray sclerae, though the expanding phenotype points to significant extra-skeletal manifestations.
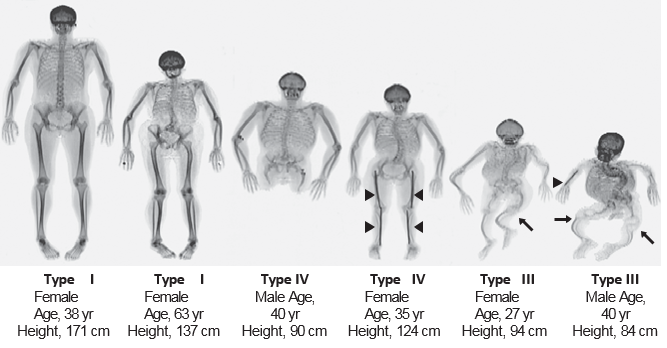
There are 13 genes known to cause OI with mutations in the type I collagen genes, COL1A1 and COL1A2 that account for 90% of mutant alleles. Biochemical and molecular mechanisms of OI include haploinsufficient type I collagen OI, dominant negative type I collagen OI, disorders of type I collagen post-translational modification, disorders of type I collagen transport, and an emerging number of disorders due to abnormal signaling. Some of the other recently discovered genes causing OI lead to distinct manifestations. For example, type V OI, due to a mutation in IFITM5, has the unique features of calcification of the interosseous membrane of the forearm and hyperplastic callus formation upon fracture. Based upon this current knowledge, it is certain that there are differences in morbidity and mortality, disease progression and response to current and emerging therapies among the different forms of OI. There at least another eight recognized “types” of OI, designated Type I through Type VIII. The types can be distinguished by their signs and symptoms, although their phenotypic features overlap. Type I is the mildest form and Type II is the most severe; the other types have features that fall somewhere between these two extremes.
Scoliosis in OI
Children with OI can develop scoliosis due to vertebral fractures, muscle hypotonia and ligamentous laxity. The prevalence of scoliosis in OI varies between 39% and 80%. Spinal curvatures in children with OI tend to progress rapidly, especially during the adolescent growth spurt. Scoliosis in OI is a significant contributor to morbidity and mortality due to reduced pulmonary forced vital capacity, impaired mobility, and reduction in bone mineral density, pain, and functional impairments (2-4).
Scoliosis is commonly associated with severe OI (5,6). A few cross-sectional studies have analyzed the relationship between scoliosis and various outcomes in OI (2,4), but longitudinal data are lacking. Key issues that are thought to be related to scoliosis are decreased pulmonary function, low mobility and decreased function. However, cross-sectional studies are not adequate to determine the cause and the effect of this relationship. For example, recent evidence suggests that diminished pulmonary function may be a direct result of the collagen mutation rather than extrinsic factors such as scoliosis and chest wall compliance. It is therefore possible that the relationship between scoliosis and low pulmonary function found in cross-sectional studies are correlative rather than causative. For example, it can be envisioned that both the scoliosis and the low pulmonary function are independent manifestations of mutations that lead to severe disease. Longitudinal studies are needed to evaluate the time course and progression of both the scoliosis and functional outcomes in individuals with OI. These considerations also have important implications for treatment planning and follow-up of children and adults with OI. Treatment of scoliosis includes observation, bracing, and surgery, but the evidence on the effects of these treatment strategies on the pulmonary function, functional ability and quality of life of children and adolescents with OI is limited.
Dental and Craniofacial Abnormalities in OI
Dental abnormalities are very frequent in OI. Dentinogenesis Imperfecta (DI) is one of the key features of the disorder and its presence or absence has been used as one of the criteria to classify individuals with OI into several OI types14. Studies at the Shriners Hospital in Montreal suggest that the presence of DI shows a strong genotype-phenotype correlation (5,6). Clinically-apparent DI is almost always absent in the presence of COL1A1 haploinsufficiency mutations (1). When OI is caused by triple helical glycine mutations, DI is usually present but depends on the location of the affected glycine residue (8). In some OI cases, the DI phenotype is only detected by radiographs.
Dental aberrations other than DI are also very common. A study on 69 children and adolescents found that tooth agenesis, apically extended pulp chambers, and impaction of second permanent molars each affected more than 20% of patients (9). Similar observations have been made in adults with OI (10). Regarding craniofacial abnormalities, a study on 59 individuals with OI observed a specific type of malocclusion in OI types III and IV (11). This malocclusion was caused by a hypoplastic maxilla and a counterclockwise rotation of the mandible, resulting in a concave profile and a reverse anterior bite. Frequently there was also a lateral open bite that was caused by deficient development of the dentoalveolar bone in the posterior segments. In OI type I, milder types of malocclusion were observed which were more similar to the general population.